
Haber process
This article's lead section may be too short to adequately summarize the key points. (November 2023) |

The Haber process,[1] also called the Haber–Bosch process, is the main industrial procedure for the production of ammonia.[2][3] It converts atmospheric nitrogen (N2) to ammonia (NH3) by a reaction with hydrogen (H2) using finely divided iron metal as a catalyst:

This reaction is thermodynamically favorable at room temperature, but the kinetics are prohibitively slow. At high temperatures at which catalysts are active enough that the reaction proceeds to equilibrium, the reaction is reactant-favored rather than product-favored. As a result, high pressures are needed to drive the reaction forward.
The German chemists Fritz Haber and Carl Bosch developed the process in the first decade of the 20th century, and its improved efficiency over existing methods such as the Birkeland-Eyde and Frank-Caro processes was a major advancement in the industrial production of ammonia.[4][5][6] The Haber process can be combined with steam reforming to produce ammonia with just three chemical inputs: water, natural gas, and atmospheric nitrogen. Both Haber and Bosch were eventually awarded the Nobel Prize in Chemistry: Haber in 1918 for ammonia synthesis specifically, and Bosch in 1931 for related contributions to high-pressure chemistry.
History
[edit].jpg)
During the 19th century, the demand rapidly increased for nitrates and ammonia for use as fertilizers, which supply plants with the nutrients they need to grow, and for industrial feedstocks. The main source was mining niter deposits and guano from tropical islands.[7] At the beginning of the 20th century these reserves were thought insufficient to satisfy future demands,[8] and research into new potential sources of ammonia increased. Although atmospheric nitrogen (N2) is abundant, comprising ~78% of the air, it is exceptionally stable and does not readily react with other chemicals.
Haber, with his assistant Robert Le Rossignol[citation needed], developed the high-pressure devices and catalysts needed to demonstrate the Haber process at a laboratory scale.[9][10] They demonstrated their process in the summer of 1909 by producing ammonia from the air, drop by drop, at the rate of about 125 mL (4 US fl oz) per hour. The process was purchased by the German chemical company BASF, which assigned Carl Bosch the task of scaling up Haber's tabletop machine to industrial scale.[5][11] He succeeded in 1910. Haber and Bosch were later awarded Nobel Prizes, in 1918 and 1931 respectively, for their work in overcoming the chemical and engineering problems of large-scale, continuous-flow, high-pressure technology.[5]
Ammonia was first manufactured using the Haber process on an industrial scale in 1913 in BASF's Oppau plant in Germany, reaching 20 tonnes/day in 1914.[12] During World War I, the production of munitions required large amounts of nitrate. The Allied powers had access to large deposits of sodium nitrate in Chile (Chile saltpetre) controlled by British companies. India had large supplies too, but it was also controlled by the British.[13] Moreover, even if German commercial interests had nominal legal control of such resources, the Allies controlled the sea lanes and imposed a highly effective blockade which would have prevented such supplies from reaching Germany. The Haber process proved so essential to the German war effort[5][14] that it is considered virtually certain Germany would have been defeated in a matter of months without it. Synthetic ammonia from the Haber process was used for the production of nitric acid, a precursor to the nitrates used in explosives.
The original Haber–Bosch reaction chambers used osmium as the catalyst, but this was available in extremely small quantities. Haber noted that uranium was almost as effective and easier to obtain than osmium. In 1909, BASF researcher Alwin Mittasch discovered a much less expensive iron-based catalyst that is still used. A major contributor to the discovery of this catalysis was Gerhard Ertl.[15][16][17][18] The most popular catalysts are based on iron promoted with K2O, CaO, SiO2, and Al2O3.
During the interwar years, alternative processes were developed, most notably the Casale process, the Claude process, and the Mont-Cenis process developed by the Friedrich Uhde Ingenieurbüro.[19] Luigi Casale and Georges Claude proposed to increase the pressure of the synthesis loop to 80–100 MPa (800–1,000 bar; 12,000–15,000 psi), thereby increasing the single-pass ammonia conversion and making nearly complete liquefaction at ambient temperature feasible. Claude proposed to have three or four converters with liquefaction steps in series, thereby avoiding recycling. Most plants continue to use the original Haber process (20 MPa (200 bar; 2,900 psi) and 500 °C (932 °F)), albeit with improved single-pass conversion and lower energy consumption due to process and catalyst optimization.
Process
[edit]
Combined with the energy needed to produce hydrogen and purified atmospheric nitrogen, ammonia production is energy-intensive, accounting for 1% to 2% of global energy consumption, 3% of global carbon emissions,[20] and 3% to 5% of natural gas consumption.[21] Hydrogen required for ammonia synthesis is most often produced through gasification of carbon-containing material, mostly natural gas, but other potential carbon sources include coal, petroleum, peat, biomass, or waste. As of 2012, the global production of ammonia produced from natural gas using the steam reforming process was 72%,[22] however in China as of 2022 natural gas and coal were responsible for 20% and 75% respectively.[23] Hydrogen can also be produced from water and electricity using electrolysis: at one time, most of Europe's ammonia was produced from the Hydro plant at Vemork. Other possibilities include biological hydrogen production or photolysis, but at present, steam reforming of natural gas is the most economical means of mass-producing hydrogen.
The choice of catalyst is important for synthesizing ammonia. In 2012, Hideo Hosono's group found that Ru-loaded calcium-aluminum oxide C12A7:e− electride works well as a catalyst and pursued more efficient formation.[24][25] This method is implemented in a small plant for ammonia synthesis in Japan.[26][27] In 2019, Hosono's group found another catalyst, a novel perovskite oxynitride-hydride BaCeO3−xNyHz, that works at lower temperature and without costly ruthenium.[28]
Hydrogen production
[edit]The major source of hydrogen is methane. Steam reforming of natural gas extracts hydrogen from methane in a high-temperature and pressure tube inside a reformer with a nickel catalyst. Other fossil fuel sources include coal, heavy fuel oil and naphtha.
Green hydrogen is produced without fossil fuels or carbon dioxide emissions from biomass, water electrolysis and thermochemical (solar or another heat source) water splitting.[29][30][31]
Starting with a natural gas (CH
4) feedstock, the steps are as follows;
- Remove sulfur compounds from the feedstock, because sulfur deactivates the catalysts used in subsequent steps. Sulfur removal requires catalytic hydrogenation to convert sulfur compounds in the feedstocks to gaseous hydrogen sulfide (hydrodesulfurization, hydrotreating):

- Hydrogen sulfide is adsorbed and removed by passing it through beds of zinc oxide where it is converted to solid zinc sulfide:


- Catalytic steam reforming of the sulfur-free feedstock forms hydrogen plus carbon monoxide:

- Catalytic shift conversion converts the carbon monoxide to carbon dioxide and more hydrogen:

- Carbon dioxide is removed either by absorption in aqueous ethanolamine solutions or by adsorption in pressure swing adsorbers (PSA) using proprietary solid adsorption media.
- The final step in producing hydrogen is to use catalytic methanation to remove residual carbon monoxide or carbon dioxide:


Ammonia production
[edit]The hydrogen is catalytically reacted with nitrogen (derived from process air[clarification needed]) to form anhydrous liquid ammonia. It is difficult and expensive, as lower temperatures result in slower reaction kinetics (hence a slower reaction rate)[32] and high pressure requires high-strength pressure vessels[33] that resist hydrogen embrittlement. Diatomic nitrogen is bound together by a triple bond, which makes it relatively inert.[34][35] Yield and efficiency are low, meaning that the ammonia must be extracted and the gases reprocessed for the reaction to proceed at an acceptable pace.[36]
This step is known as the ammonia synthesis loop:

The gases (nitrogen and hydrogen) are passed over four beds of catalyst, with cooling between each pass to maintain a reasonable equilibrium constant. On each pass, only about 15% conversion occurs, but unreacted gases are recycled, and eventually conversion of 97% is achieved.[3]
Due to the nature of the (typically multi-promoted magnetite) catalyst used in the ammonia synthesis reaction, only low levels of oxygen-containing (especially CO, CO2 and H2O) compounds can be tolerated in the hydrogen/nitrogen mixture. Relatively pure nitrogen can be obtained by air separation, but additional oxygen removal may be required.
Because of relatively low single pass conversion rates (typically less than 20%), a large recycle stream is required. This can lead to the accumulation of inerts in the gas.
Nitrogen gas (N2) is unreactive because the atoms are held together by triple bonds. The Haber process relies on catalysts that accelerate the scission of these bonds.
Two opposing considerations are relevant: the equilibrium position and the reaction rate. At room temperature, the equilibrium is in favor of ammonia, but the reaction does not proceed at a detectable rate due to its high activation energy. Because the reaction is exothermic, the equilibrium constant decreases with increasing temperature following Le Châtelier's principle. It becomes unity at around 150–200 °C (302–392 °F).[3]
| Temperature (°C) |
Kp [clarification needed] |
|---|---|
| 300 | 4.34 × 10−3 |
| 400 | 1.64 × 10−4 |
| 450 | 4.51 × 10−5 |
| 500 | 1.45 × 10−5 |
| 550 | 5.38 × 10−6 |
| 600 | 2.25 × 10−6 |
Above this temperature, the equilibrium quickly becomes unfavorable at atmospheric pressure, according to the Van 't Hoff equation. Lowering the temperature is unhelpful because the catalyst requires a temperature of at least 400 °C to be efficient.[3]
Increased pressure favors the forward reaction because 4 moles of reactant produce 2 moles of product, and the pressure used (15–25 MPa (150–250 bar; 2,200–3,600 psi)) alters the equilibrium concentrations to give a substantial ammonia yield. The reason for this is evident in the equilibrium relationship:

where is the fugacity coefficient of species , is the mole fraction of the same species, is the reactor pressure, and is standard pressure, typically 1 bar (0.10 MPa).





Economically, reactor pressurization is expensive: pipes, valves, and reaction vessels need to be strong enough, and safety considerations affect operating at 20 MPa. Compressors take considerable energy, as work must be done on the (compressible) gas. Thus, the compromise used gives a single-pass yield of around 15%.[3]
While removing the ammonia from the system increases the reaction yield, this step is not used in practice, since the temperature is too high; instead it is removed from the gases leaving the reaction vessel. The hot gases are cooled under high pressure, allowing the ammonia to condense and be removed as a liquid. Unreacted hydrogen and nitrogen gases are returned to the reaction vessel for another round.[3] While most ammonia is removed (typically down to 2–5 mol.%), some ammonia remains in the recycle stream. In academic literature, a more complete separation of ammonia has been proposed by absorption in metal halides, metal-organic frameworks or zeolites.[38] Such a process is called an absorbent-enhanced Haber process or adsorbent-enhanced Haber–Bosch process.[39]
Pressure/temperature
[edit]The steam reforming, shift conversion, carbon dioxide removal, and methanation steps each operate at absolute pressures of about 25 to 35 bar, while the ammonia synthesis loop operates at temperatures of 300–500 °C (572–932 °F) and pressures ranging from 60 to 180 bar depending upon the method used. The resulting ammonia must then be separated from the residual hydrogen and nitrogen at temperatures of −20 °C (−4 °F).[40][3]
Catalysts
[edit].jpg)

The Haber–Bosch process relies on catalysts to accelerate N2 hydrogenation. The catalysts are heterogeneous solids that interact with gaseous reagents.[41]
The catalyst typically consists of finely divided iron bound to an iron oxide carrier containing promoters possibly including aluminium oxide, potassium oxide, calcium oxide, potassium hydroxide,[42] molybdenum,[43] and magnesium oxide.
Iron-based catalysts
[edit]The iron catalyst is obtained from finely ground iron powder, which is usually obtained by reduction of high-purity magnetite (Fe3O4). The pulverized iron is oxidized to give magnetite or wüstite (FeO, ferrous oxide) particles of a specific size. The magnetite (or wüstite) particles are then partially reduced, removing some of the oxygen. The resulting catalyst particles consist of a core of magnetite, encased in a shell of wüstite, which in turn is surrounded by an outer shell of metallic iron. The catalyst maintains most of its bulk volume during the reduction, resulting in a highly porous high-surface-area material, which enhances its catalytic effectiveness. Minor components include calcium and aluminium oxides, which support the iron catalyst and help it maintain its surface area. These oxides of Ca, Al, K, and Si are unreactive to reduction by hydrogen.[3]
The production of the catalyst requires a particular melting process in which used raw materials must be free of catalyst poisons and the promoter aggregates must be evenly distributed in the magnetite melt. Rapid cooling of the magnetite, which has an initial temperature of about 3500 °C, produces the desired precursor. Unfortunately, the rapid cooling ultimately forms a catalyst of reduced abrasion resistance. Despite this disadvantage, the method of rapid cooling is often employed.[3]
The reduction of the precursor magnetite to α-iron is carried out directly in the production plant with synthesis gas. The reduction of the magnetite proceeds via the formation of wüstite (FeO) so that particles with a core of magnetite become surrounded by a shell of wüstite. The further reduction of magnetite and wüstite leads to the formation of α-iron, which forms together with the promoters the outer shell.[44] The involved processes are complex and depend on the reduction temperature: At lower temperatures, wüstite disproportionates into an iron phase and a magnetite phase; at higher temperatures, the reduction of the wüstite and magnetite to iron dominates.[45]
The α-iron forms primary crystallites with a diameter of about 30 nanometers. These crystallites form a bimodal pore system with pore diameters of about 10 nanometers (produced by the reduction of the magnetite phase) and of 25 to 50 nanometers (produced by the reduction of the wüstite phase).[44] With the exception of cobalt oxide, the promoters are not reduced.
During the reduction of the iron oxide with synthesis gas, water vapor is formed. This water vapor must be considered for high catalyst quality as contact with the finely divided iron would lead to premature aging of the catalyst through recrystallization, especially in conjunction with high temperatures. The vapor pressure of the water in the gas mixture produced during catalyst formation is thus kept as low as possible, target values are below 3 gm−3. For this reason, the reduction is carried out at high gas exchange, low pressure, and low temperatures. The exothermic nature of the ammonia formation ensures a gradual increase in temperature.[3]
The reduction of fresh, fully oxidized catalyst or precursor to full production capacity takes four to ten days.[3] The wüstite phase is reduced faster and at lower temperatures than the magnetite phase (Fe3O4). After detailed kinetic, microscopic, and X-ray spectroscopic investigations it was shown that wüstite reacts first to metallic iron. This leads to a gradient of iron(II) ions, whereby these diffuse from the magnetite through the wüstite to the particle surface and precipitate there as iron nuclei. A high-activity novel catalyst based on this phenomenon was discovered in the 1980s at the Zhejiang University of Technology and commercialized by 2003.[46]
Pre-reduced, stabilized catalysts occupy a significant market share. They are delivered showing the fully developed pore structure, but have been oxidized again on the surface after manufacture and are therefore no longer pyrophoric. The reactivation of such pre-reduced catalysts requires only 30 to 40 hours instead of several days. In addition to the short start-up time, they have other advantages such as higher water resistance and lower weight.[3]
| Typical catalyst composition[47] | Iron (%) | Potassium (%) | Aluminium (%) | Calcium (%) | Oxygen (%) |
|---|---|---|---|---|---|
| Volume composition | 40.5 | 0.35 | 2.0 | 1.7 | 53.2 |
| Surface composition before reduction | 8.6 | 36.1 | 10.7 | 4.7 | 40.0 |
| Surface composition after reduction | 11.0 | 27.0 | 17.0 | 4.0 | 41.0 |
Catalysts other than iron
[edit]Many efforts have been made to improve the Haber–Bosch process. Many metals were tested as catalysts. The requirement for suitability is the dissociative adsorption of nitrogen (i. e. the nitrogen molecule must be split into nitrogen atoms upon adsorption). If the binding of the nitrogen is too strong, the catalyst is blocked and the catalytic ability is reduced (self-poisoning). The elements in the periodic table to the left of the iron group show such strong bonds. Further, the formation of surface nitrides makes, for example, chromium catalysts ineffective. Metals to the right of the iron group, in contrast, adsorb nitrogen too weakly for ammonia synthesis. Haber initially used catalysts based on osmium and uranium. Uranium reacts to its nitride during catalysis, while osmium oxide is rare.[48]
According to theoretical and practical studies, improvements over pure iron are limited. The activity of iron catalysts is increased by the inclusion of cobalt.[49]
Ruthenium
[edit]Ruthenium forms highly active catalysts. Allowing milder operating pressures and temperatures, Ru-based materials are referred to as second-generation catalysts. Such catalysts are prepared by the decomposition of triruthenium dodecacarbonyl on graphite.[3] A drawback of activated-carbon-supported ruthenium-based catalysts is the methanation of the support in the presence of hydrogen. Their activity is strongly dependent on the catalyst carrier and the promoters. A wide range of substances can be used as carriers, including carbon, magnesium oxide, aluminium oxide, zeolites, spinels, and boron nitride.[50]
Ruthenium-activated carbon-based catalysts have been used industrially in the KBR Advanced Ammonia Process (KAAP) since 1992.[51] The carbon carrier is partially degraded to methane; however, this can be mitigated by a special treatment of the carbon at 1500 °C, thus prolonging the catalyst lifetime. In addition, the finely dispersed carbon poses a risk of explosion. For these reasons and due to its low acidity, magnesium oxide has proven to be a good choice of carrier. Carriers with acidic properties extract electrons from ruthenium, make it less reactive, and have the undesirable effect of binding ammonia to the surface.[50]
Catalyst poisons
[edit]Catalyst poisons lower catalyst activity. They are usually impurities in the synthesis gas. Permanent poisons cause irreversible loss of catalytic activity, while temporary poisons lower the activity while present. Sulfur compounds, phosphorus compounds, arsenic compounds, and chlorine compounds are permanent poisons. Oxygenic compounds like water, carbon monoxide, carbon dioxide, and oxygen are temporary poisons.[3][52]
Although chemically inert components of the synthesis gas mixture such as noble gases or methane are not strictly poisons, they accumulate through the recycling of the process gases and thus lower the partial pressure of the reactants, which in turn slows conversion.[53]
Industrial production
[edit]Synthesis parameters
[edit]| temperature (°C) | Keq |
|---|---|
| 300 | 4.34 × 10−3 |
| 400 | 1.64 × 10−4 |
| 450 | 4.51 × 10−5 |
| 500 | 1.45 × 10−5 |
| 550 | 5.38 × 10−6 |
| 600 | 2.25 × 10−6 |
The reaction is:

The reaction is an exothermic equilibrium reaction in which the gas volume is reduced. The equilibrium constant Keq of the reaction (see table) and obtained from:

Since the reaction is exothermic, the equilibrium of the reaction shifts at lower temperatures to the ammonia side. Furthermore, four volumetric units of the raw materials produce two volumetric units of ammonia. According to Le Chatelier's principle, higher pressure favours ammonia. High pressure is necessary to ensure sufficient surface coverage of the catalyst with nitrogen.[56] For this reason, a ratio of nitrogen to hydrogen of 1 to 3, a pressure of 250 to 350 bar, a temperature of 450 to 550 °C and α iron are optimal.
The catalyst ferrite (α-Fe) is produced in the reactor by the reduction of magnetite with hydrogen. The catalyst has its highest efficiency at temperatures of about 400 to 500 °C. Even though the catalyst greatly lowers the activation energy for the cleavage of the triple bond of the nitrogen molecule, high temperatures are still required for an appropriate reaction rate. At the industrially used reaction temperature of 450 to 550 °C an optimum between the decomposition of ammonia into the starting materials and the effectiveness of the catalyst is achieved.[57] The formed ammonia is continuously removed from the system. The volume fraction of ammonia in the gas mixture is about 20%.
The inert components, especially the noble gases such as argon, should not exceed a certain content in order not to reduce the partial pressure of the reactants too much. To remove the inert gas components, part of the gas is removed and the argon is separated in a gas separation plant. The extraction of pure argon from the circulating gas is carried out using the Linde process.[58]
Large-scale implementation
[edit]Modern ammonia plants produce more than 3000 tons per day in one production line. The following diagram shows the set-up of a Haber–Bosch plant:

Depending on its origin, the synthesis gas must first be freed from impurities such as hydrogen sulfide or organic sulphur compounds, which act as a catalyst poison. High concentrations of hydrogen sulfide, which occur in synthesis gas from carbonization coke, are removed in a wet cleaning stage such as the sulfosolvan process, while low concentrations are removed by adsorption on activated carbon.[59] Organosulfur compounds are separated by pressure swing adsorption together with carbon dioxide after CO conversion.
To produce hydrogen by steam reforming, methane reacts with water vapor using a nickel oxide-alumina catalyst in the primary reformer to form carbon monoxide and hydrogen. The energy required for this, the enthalpy ΔH, is 206 kJ/mol.[60]

The methane gas reacts in the primary reformer only partially. To increase the hydrogen yield and keep the content of inert components (i. e. methane) as low as possible, the remaining methane gas is converted in a second step with oxygen to hydrogen and carbon monoxide in the secondary reformer. The secondary reformer is supplied with air as the oxygen source. Also, the required nitrogen for the subsequent ammonia synthesis is added to the gas mixture.

In the third step, the carbon monoxide is oxidized to carbon dioxide, which is called CO conversion or water-gas shift reaction.

Carbon monoxide and carbon dioxide would form carbamates with ammonia, which would clog (as solids) pipelines and apparatus within a short time. In the following process step, the carbon dioxide must therefore be removed from the gas mixture. In contrast to carbon monoxide, carbon dioxide can easily be removed from the gas mixture by gas scrubbing with triethanolamine. The gas mixture then still contains methane and noble gases such as argon, which, however, behave inertly.[53]
The gas mixture is then compressed to operating pressure by turbo compressors. The resulting compression heat is dissipated by heat exchangers; it is used to preheat raw gases.
The actual production of ammonia takes place in the ammonia reactor. The first reactors were bursting under high pressure because the atomic hydrogen in the carbonaceous steel partially recombined into methane and produced cracks in the steel. Bosch, therefore, developed tube reactors consisting of a pressure-bearing steel tube in which a low-carbon iron lining tube was inserted and filled with the catalyst. Hydrogen that diffused through the inner steel pipe escaped to the outside via thin holes in the outer steel jacket, the so-called Bosch holes.[55] A disadvantage of the tubular reactors was the relatively high-pressure loss, which had to be applied again by compression. The development of hydrogen-resistant chromium-molybdenum steels made it possible to construct single-walled pipes.[61]

Modern ammonia reactors are designed as multi-storey reactors with a low-pressure drop, in which the catalysts are distributed as fills over about ten storeys one above the other. The gas mixture flows through them one after the other from top to bottom. Cold gas is injected from the side for cooling. A disadvantage of this reactor type is the incomplete conversion of the cold gas mixture in the last catalyst bed.[61]
Alternatively, the reaction mixture between the catalyst layers is cooled using heat exchangers, whereby the hydrogen-nitrogen mixture is preheated to the reaction temperature. Reactors of this type have three catalyst beds. In addition to good temperature control, this reactor type has the advantage of better conversion of the raw material gases compared to reactors with cold gas injection.
Uhde has developed and is using an ammonia converter with three radial flow catalyst beds and two internal heat exchangers instead of axial flow catalyst beds. This further reduces the pressure drop in the converter.[62]
The reaction product is continuously removed for maximum yield. The gas mixture is cooled to 450 °C in a heat exchanger using water, freshly supplied gases, and other process streams. The ammonia also condenses and is separated in a pressure separator. Unreacted nitrogen and hydrogen are then compressed back to the process by a circulating gas compressor, supplemented with fresh gas, and fed to the reactor.[61] In a subsequent distillation, the product ammonia is purified.
Mechanism
[edit]Elementary steps
[edit]The mechanism of ammonia synthesis contains the following seven elementary steps:
- transport of the reactants from the gas phase through the boundary layer to the surface of the catalyst.
- pore diffusion to the reaction center
- adsorption of reactants
- reaction
- desorption of product
- transport of the product through the pore system back to the surface
- transport of the product into the gas phase
Transport and diffusion (the first and last two steps) are fast compared to adsorption, reaction, and desorption because of the shell structure of the catalyst. It is known from various investigations that the rate-determining step of the ammonia synthesis is the dissociation of nitrogen.[3] In contrast, exchange reactions between hydrogen and deuterium on the Haber–Bosch catalysts still take place at temperatures of −196 °C (−320.8 °F) at a measurable rate; the exchange between deuterium and hydrogen on the ammonia molecule also takes place at room temperature. Since the adsorption of both molecules is rapid, it cannot determine the speed of ammonia synthesis.[63]
In addition to the reaction conditions, the adsorption of nitrogen on the catalyst surface depends on the microscopic structure of the catalyst surface. Iron has different crystal surfaces, whose reactivity is very different. The Fe(111) and Fe(211) surfaces have by far the highest activity. The explanation for this is that only these surfaces have so-called C7 sites – these are iron atoms with seven closest neighbours.[3]
The dissociative adsorption of nitrogen on the surface follows the following scheme, where S* symbolizes an iron atom on the surface of the catalyst:[44]
- N2 → S*–N2 (γ-species) → S*–N2–S* (α-species) → 2 S*–N (β-species, surface nitride)
The adsorption of nitrogen is similar to the chemisorption of carbon monoxide. On a Fe(111) surface, the adsorption of nitrogen first leads to an adsorbed γ-species with an adsorption energy of 24 kJmol−1 and an N-N stretch vibration of 2100 cm−1. Since the nitrogen is isoelectronic to carbon monoxide, it adsorbs in an on-end configuration in which the molecule is bound perpendicular to the metal surface at one nitrogen atom.[17][64][3] This has been confirmed by photoelectron spectroscopy.[65]
Ab-initio-MO calculations have shown that, in addition to the σ binding of the free electron pair of nitrogen to the metal, there is a π binding from the d orbitals of the metal to the π* orbitals of nitrogen, which strengthens the iron-nitrogen bond. The nitrogen in the α state is more strongly bound with 31 kJmol−1. The resulting N–N bond weakening could be experimentally confirmed by a reduction of the wave numbers of the N–N stretching oscillation to 1490 cm−1.[64]
Further heating of the Fe(111) area covered by α-N2 leads to both desorption and the emergence of a new band at 450 cm−1. This represents a metal-nitrogen oscillation, the β state. A comparison with vibration spectra of complex compounds allows the conclusion that the N2 molecule is bound "side-on", with an N atom in contact with a C7 site. This structure is called "surface nitride". The surface nitride is very strongly bound to the surface.[65] Hydrogen atoms (Hads), which are very mobile on the catalyst surface, quickly combine with it.
Infrared spectroscopically detected surface imides (NHad), surface amides (NH2,ad) and surface ammoniacates (NH3,ad) are formed, the latter decay under NH3 release (desorption).[55] The individual molecules were identified or assigned by X-ray photoelectron spectroscopy (XPS), high-resolution electron energy loss spectroscopy (HREELS) and Ir Spectroscopy.
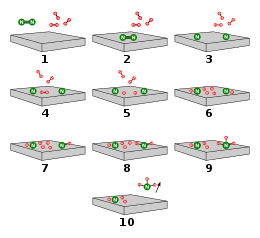
Drawn reaction scheme

On the basis of these experimental findings, the reaction mechanism is believed to involve the following steps (see also figure):[66]
- N2 (g) → N2 (adsorbed)
- N2 (adsorbed) → 2 N (adsorbed)
- H2 (g) → H2 (adsorbed)
- H2 (adsorbed) → 2 H (adsorbed)
- N (adsorbed) + 3 H (adsorbed) → NH3 (adsorbed)
- NH3 (adsorbed) → NH3 (g)
Reaction 5 occurs in three steps, forming NH, NH2, and then NH3. Experimental evidence points to reaction 2 as being slow, rate-determining step. This is not unexpected, since that step breaks the nitrogen triple bond, the strongest of the bonds broken in the process.
As with all Haber–Bosch catalysts, nitrogen dissociation is the rate-determining step for ruthenium-activated carbon catalysts. The active center for ruthenium is a so-called B5 site, a 5-fold coordinated position on the Ru(0001) surface where two ruthenium atoms form a step edge with three ruthenium atoms on the Ru(0001) surface.[67] The number of B5 sites depends on the size and shape of the ruthenium particles, the ruthenium precursor and the amount of ruthenium used.[50] The reinforcing effect of the basic carrier used in the ruthenium catalyst is similar to the promoter effect of alkali metals used in the iron catalyst.[50]
Energy diagram
[edit]
An energy diagram can be created based on the Enthalpy of Reaction of the individual steps. The energy diagram can be used to compare homogeneous and heterogeneous reactions: Due to the high activation energy of the dissociation of nitrogen, the homogeneous gas phase reaction is not realizable. The catalyst avoids this problem as the energy gain resulting from the binding of nitrogen atoms to the catalyst surface overcompensates for the necessary dissociation energy so that the reaction is finally exothermic. Nevertheless, the dissociative adsorption of nitrogen remains the rate-determining step: not because of the activation energy, but mainly because of the unfavorable pre-exponential factor of the rate constant. Although hydrogenation is endothermic, this energy can easily be applied by the reaction temperature (about 700 K).[3]
Economic and environmental aspects
[edit]| External videos | |
|---|---|

When first invented, the Haber process competed against another industrial process, the cyanamide process. However, the cyanamide process consumed large amounts of electrical power and was more labor-intensive than the Haber process.[5]: 137–143
As of 2018, the Haber process produces 230 million tonnes of anhydrous ammonia per year.[68] The ammonia is used mainly as a nitrogen fertilizer as ammonia itself, in the form of ammonium nitrate, and as urea. The Haber process consumes 3–5% of the world's natural gas production (around 1–2% of the world's energy supply).[4][69][70][71] In combination with advances in breeding, herbicides, and pesticides, these fertilizers have helped to increase the productivity of agricultural land:
With average crop yields remaining at the 1900 level the crop harvest in the year 2000 would have required nearly four times more land and the cultivated area would have claimed nearly half of all ice-free continents, rather than under 15% of the total land area that is required today.[72]
— Vaclav Smil, Nitrogen cycle and world food production, Volume 2, pages 9–13
The energy-intensity of the process contributes to climate change and other environmental problems such as the leaching of nitrates into groundwater, rivers, ponds, and lakes; expanding dead zones in coastal ocean waters, resulting from recurrent eutrophication; atmospheric deposition of nitrates and ammonia affecting natural ecosystems; higher emissions of nitrous oxide (N2O), now the third most important greenhouse gas following CO2 and CH4.[72] The Haber–Bosch process is one of the largest contributors to a buildup of reactive nitrogen in the biosphere, causing an anthropogenic disruption to the nitrogen cycle.[73]
Since nitrogen use efficiency is typically less than 50%,[74] farm runoff from heavy use of fixed industrial nitrogen disrupts biological habitats.[4][75]
Nearly 50% of the nitrogen found in human tissues originated from the Haber–Bosch process.[76] Thus, the Haber process serves as the "detonator of the population explosion", enabling the global population to increase from 1.6 billion in 1900 to 7.7 billion by November 2018.[77]
Reverse fuel cell[78] technology converts electric energy, water and nitrogen into ammonia without a separate hydrogen electrolysis process.[79]
The use of synthetic nitrogen fertilisers reduces the incentive for farmers to use more sustainable crop rotations which include legumes for their natural nitrogen-fixing ability.
See also
[edit]- Birkeland–Eyde process – Nitrogen fixation process using electrical arcs
- Calcium cyanamide – a calcium salt of the cyanamide which is used as plant fertilizer
- Chilean saltpeter – Mineral form of sodium nitrate
- Guano – Excrement of seabirds or bats
- Hydrogen production – Industrial production of molecular hydrogen
- Industrial gas – Gaseous materials produced for use in industry
- Paradas method – Process to extract nitrate from caliche by leaching
- Crop rotation
- Legume
References
[edit]- ^ Habers process chemistry. India: Arihant publications. 2018. p. 264. ISBN 978-93-131-6303-9.
- ^ Appl, M. (1982). "The Haber–Bosch Process and the Development of Chemical Engineering". A Century of Chemical Engineering. New York: Plenum Press. pp. 29–54. ISBN 978-0-306-40895-3.
- ^ a b c d e f g h i j k l m n o p q r Appl, Max (2006). "Ammonia". Ullmann's Encyclopedia of Industrial Chemistry. Weinheim: Wiley-VCH. doi:10.1002/14356007.a02_143.pub2. ISBN 978-3527306732.
- ^ a b c Smil, Vaclav (2004). Enriching the Earth: Fritz Haber, Carl Bosch, and the Transformation of World Food Production (1st ed.). Cambridge, MA: MIT. ISBN 978-0-262-69313-4.
- ^ a b c d e Hager, Thomas (2008). The Alchemy of Air: A Jewish genius, a doomed tycoon, and the scientific discovery that fed the world but fueled the rise of Hitler (1st ed.). New York, New York: Harmony Books. ISBN 978-0-307-35178-4.
- ^ Sittig, Marshall (1979). Fertilizer Industry: Processes, Pollution Control, and Energy Conservation. Park Ridge, New Jersey: Noyes Data Corp. ISBN 978-0-8155-0734-5.
- ^ Vandermeer, John (2011). The Ecology of Agroecosystems. Jones & Bartlett Learning. p. 149. ISBN 978-0-7637-7153-9.
- ^ James, Laylin K. (1993). Nobel Laureates in Chemistry 1901–1992 (3rd ed.). Washington, DC: American Chemical Society. p. 118. ISBN 978-0-8412-2690-6.
- ^ Haber, Fritz (1905). Thermodynamik technischer Gasreaktionen (in German) (1st ed.). Paderborn: Salzwasser Verlag. ISBN 978-3-86444-842-3.
- ^ "Robert Le Rossignol, 1884–1976: Professional Chemist" (PDF), ChemUCL Newsletter, p. 8, 2009, archived from the original (PDF) on 13 January 2011.
- ^ Bosch, Carl (2 March 1908). U.S. patent 990,191.
- ^ Philip, Phylis Morrison (2001). "Fertile Minds (Book Review of Enriching the Earth: Fritz Haber, Carl Bosch, and the Transformation of World Food Production)". American Scientist. Archived from the original on 2 July 2012.
- ^ Brown, GI (2011). Explosives: History with a Bang (1 ed.). The History Press. ISBN 978-0752456966.
- ^ "Nobel Award to Haber" (PDF). The New York Times. 3 February 1920. Archived from the original (PDF) on 24 February 2021. Retrieved 11 October 2010.
- ^ Bozso, F.; Ertl, G.; Grunze, M.; Weiss, M. (1977). "Interaction of nitrogen with iron surfaces: I. Fe(100) and Fe(111)". Journal of Catalysis. 49 (1): 18–41. doi:10.1016/0021-9517(77)90237-8.
- ^ Imbihl, R.; Behm, R. J.; Ertl, G.; Moritz, W. (1982). "The structure of atomic nitrogen adsorbed on Fe(100)" (PDF). Surface Science. 123 (1): 129–140. Bibcode:1982SurSc.123..129I. doi:10.1016/0039-6028(82)90135-2.
- ^ a b Ertl, G.; Lee, S. B.; Weiss, M. (1982). "Kinetics of nitrogen adsorption on Fe(111)". Surface Science. 114 (2–3): 515–526. Bibcode:1982SurSc.114..515E. doi:10.1016/0039-6028(82)90702-6.
- ^ Ertl, G. (1983). "Primary steps in catalytic synthesis of ammonia". Journal of Vacuum Science and Technology A. 1 (2): 1247–1253. Bibcode:1983JVSTA...1.1247E. doi:10.1116/1.572299.
- ^ "100 years of Thyssenkrupp Uhde". Industrial Solutions (in German). Retrieved 8 December 2021.
- ^ "Electrochemically-produced ammonia could revolutionize food production". 9 July 2018. Retrieved 15 December 2018.
Ammonia manufacturing consumes 1 to 2% of total global energy and is responsible for approximately 3% of global carbon dioxide emissions.
- ^ Song, Yang; Hensley, Dale; Bonnesen, Peter; Liang, Liango; Huang, Jingsong; Baddorf, Arthur; Tschaplinski, Timothy; Engle, Nancy; Wu, Zili; Cullen, David; Meyer, Harry III; Sumpter, Bobby; Rondinone, Adam (2 May 2018). "A physical catalyst for the electrolysis of nitrogen to ammonia". Science Advances. 4 (4). Oak Ridge National Laboratory: e1700336. Bibcode:2018SciA....4..336S. doi:10.1126/sciadv.1700336. PMC 5922794. PMID 29719860. Retrieved 15 December 2018.
Ammonia synthesis consumes 3 to 5% of the world's natural gas, making it a significant contributor to greenhouse gas emissions.
- ^ "Ammonia". Industrial Efficiency Technology & Measures. 30 April 2013. Archived from the original on 2 October 2019. Retrieved 6 April 2018.
- ^ Zhao, Fu; Fan, Ying; Zhang, Shaohui; Eichhammer, Wolfgang; Haendel, Michael; Yu, Songmin (1 April 2022). "Exploring pathways to deep de-carbonization and the associated environmental impact in China's ammonia industry". Environmental Research Letters. 17 (4): 045029. Bibcode:2022ERL....17d5029Z. doi:10.1088/1748-9326/ac614a. ISSN 1748-9326.
- ^ Kuganathan, Navaratnarajah; Hosono, Hideo; Shluger, Alexander L.; Sushko, Peter V. (January 2014). "Enhanced N2 Dissociation on Ru-Loaded Inorganic Electride". Journal of the American Chemical Society. 136 (6): 2216–2219. doi:10.1021/ja410925g. PMID 24483141.
- ^ Hara, Michikazu; Kitano, Masaaki; Hosono, Hideo; Sushko, Peter V. (2017). "Ru-Loaded C12A7:e– Electride as a Catalyst for Ammonia Synthesi". ACS Catalysis. 7 (4): 2313–2324. doi:10.1021/acscatal.6b03357.
- ^ "Ajinomoto Co., Inc., UMI, and Tokyo Institute of Technology Professors Establish New Company to Implement the World's First On Site Production of Ammonia". Ajinomoto. 27 April 2017. Retrieved 9 November 2021.
- ^ Crolius, Stephen H. (17 December 2020). "Tsubame BHB Launches Joint Evaluation with Mitsubishi Chemical". Ammonia Energy Association. Retrieved 9 November 2021.
- ^ Kitano, Masaaki; Kujirai, Jun; Ogasawara, Kiya; Matsuishi, Satoru; Tada, Tomofumi; Abe, Hitoshi; Niwa, Yasuhiro; Hosono, Hideo (2019). "Low-Temperature Synthesis of Perovskite Oxynitride-Hydrides as Ammonia Synthesis Catalysts". Journal of the American Chemical Society. 141 (51): 20344–20353. doi:10.1021/jacs.9b10726. PMID 31755269. S2CID 208227325.
- ^ Wang, Ying; Meyer, Thomas J. (14 March 2019). "A Route to Renewable Energy Triggered by the Haber–Bosch Process". Chem. 5 (3): 496–497. Bibcode:2019Chem....5..496W. doi:10.1016/j.chempr.2019.02.021. S2CID 134713643.
- ^ Schneider, Stefan; Bajohr, Siegfried; Graf, Frank; Kolb, Thomas (13 January 2020). "State of the Art of Hydrogen Production via Pyrolysis of Natural Gas". ChemBioEng Reviews. 7 (5): 150–158. doi:10.1002/cben.202000014. S2CID 221708661 – via Wiley Online Library.
- ^ "Progress in the Electrochemical Synthesis of Ammonia | Request PDF".
- ^ Clark 2013, However, 400–450 °C isn't a low temperature! Rate considerations: The lower the temperature you use, the slower the reaction becomes. A manufacturer is trying to produce as much ammonia as possible per day. It makes no sense to try to achieve an equilibrium mixture which contains a very high proportion of ammonia if it takes several years for the reaction to reach that equilibrium"..
- ^ Clark 2013, "Rate considerations: Increasing the pressure brings the molecules closer together. In this particular instance, it will increase their chances of hitting and sticking to the surface of the catalyst where they can react. The higher the pressure the better in terms of the rate of a gas reaction. Economic considerations: Very high pressures are expensive to produce on two counts. Extremely strong pipes and containment vessels are needed to withstand the very high pressure. That increases capital costs when the plant is built".
- ^ Zhang, Xiaoping; Su, Rui; Li, Jingling; Huang, Liping; Yang, Wenwen; Chingin, Konstantin; Balabin, Roman; Wang, Jingjing; Zhang, Xinglei; Zhu, Weifeng; Huang, Keke; Feng, Shouhua; Chen, Huanwen (2024). "Efficient catalyst-free N2 fixation by water radical cations under ambient conditions". Nature Communications. 15 (1) 1535: 1535. Bibcode:2024NatCo..15.1535Z. doi:10.1038/s41467-024-45832-9. PMC 10879522. PMID 38378822.
- ^ "Chemistry of Nitrogen". Compounds. Chem.LibreTexts.org. 5 June 2019. Retrieved 7 July 2019.
- ^ Clark 2013, "At each pass of the gases through the reactor, only about 15% of the nitrogen and hydrogen converts to ammonia. (This figure also varies from plant to plant.) By continual recycling of the unreacted nitrogen and hydrogen, the overall conversion is about 98%".
- ^ Brown, Theodore L.; LeMay, H. Eugene Jr.; Bursten, Bruce E. (2006). "Table 15.2". Chemistry: The Central Science (10th ed.). Upper Saddle River, NJ: Pearson. ISBN 978-0-13-109686-8.
- ^ De Alwis Jayasinghe, Dukula; Chen, Yinlin; Li, Jiangnan; Rogacka, Justyna M.; Kippax-Jones, Meredydd; Lu, Wanpeng; Sapchenko, Sergei; Yang, Jinyue; Chansai, Sarayute; Zhou, Tianze; Guo, Lixia; Ma, Yujie; Dong, Longzhang; Polyukhov, Daniil; Shan, Lutong; Han, Yu; Crawshaw, Danielle; Zeng, Xiangdi; Zhu, Zhaodong; Hughes, Lewis; Frogley, Mark D.; Manuel, Pascal; Rudić, Svemir; Cheng, Yongqiang; Hardacre, Christopher; Schröder, Martin; Yang, Sihai (8 November 2024). "A Flexible Phosphonate Metal–Organic Framework for Enhanced Cooperative Ammonia Capture". Journal of the American Chemical Society. 146: 32040–32048. doi:10.1021/jacs.4c12430.
- ^ Abild-pedersen, Frank; Bligaard, Thomas (1 January 2014). "Exploring the limits: A low-pressure, low-temperature Haber–Bosch process". Chemical Physics Letters. 598: 108. Bibcode:2014CPL...598..108V. doi:10.1016/j.cplett.2014.03.003 – via academia.edu.
- ^ Koop, Fermin (13 January 2023). "Green ammonia (and fertilizer) may finally be in sight -- and it would be huge". ZME Science. Retrieved 21 March 2023.
- ^ Mittasch, Alwin (1926). "Bemerkungen zur Katalyse". Berichte der Deutschen Chemischen Gesellschaft (A and B Series). 59: 13–36. doi:10.1002/cber.19260590103.
- ^ "3.1 Ammonia synthesis". resources.schoolscience.co.uk. Archived from the original on 6 July 2020.
- ^ Rock, Peter A. (19 June 2013). Chemical Thermodynamics. University Science Books. p. 317. ISBN 978-1-891389-32-0.
- ^ a b c Max Appl (2006). "Ammonia". Ullmann's Encyclopedia of Industrial Chemistry. Weinheim: Wiley-VCH. doi:10.1002/14356007.a02_143.pub2. ISBN 978-3527306732.
- ^ Jozwiak, W. K.; Kaczmarek, E. (2007). "Reduction behavior of iron oxides in hydrogen and carbon monoxide atmospheres". Applied Catalysis A: General. 326: 17–27. doi:10.1016/j.apcata.2007.03.021.
- ^ Liu, Huazhang; Han, Wenfeng; Huo, Chao; Cen, Yaqing (15 September 2020). "Development and application of wüstite-based ammonia synthesis catalysts". Catalysis Today. SI: Energy and the Environment. 355: 110–127. doi:10.1016/j.cattod.2019.10.031. ISSN 0920-5861.
- ^ Ertl, Gerhard (1983). "Zum Mechanismus der Ammoniak-Synthese". Nachrichten aus Chemie, Technik und Laboratorium (in German). 31 (3): 178–182. doi:10.1002/nadc.19830310307.
- ^ Bowker, Michael (1993). "Chapter 7". In King, D. A.; Woodruff, D. P. (eds.). The Chemical Physics of Solid Surfaces. Vol. 6: Coadsorption, promoters and poisons. Elsevier. pp. 225–268. ISBN 978-0-444-81468-5.
- ^ Tavasoli, Ahmad; Trépanier, Mariane; Malek Abbaslou, Reza M.; Dalai, Ajay K.; Abatzoglou, Nicolas (1 December 2009). "Fischer–Tropsch synthesis on mono- and bimetallic Co and Fe catalysts supported on carbon nanotubes". Fuel Processing Technology. 90 (12): 1486–1494. Bibcode:2009FuPrT..90.1486T. doi:10.1016/j.fuproc.2009.07.007. ISSN 0378-3820.
- ^ a b c d You, Zhixiong; Inazu, Koji; Aika, Ken-ichi; Baba, Toshihide (October 2007). "Electronic and structural promotion of barium hexaaluminate as a ruthenium catalyst support for ammonia synthesis". Journal of Catalysis. 251 (2): 321–331. doi:10.1016/j.jcat.2007.08.006.
- ^ Rosowski, F.; Hornung, A.; Hinrichsen, O.; Herein, D.; Muhler, M. (April 1997). "Ruthenium catalysts for ammonia synthesis at high pressures: Preparation, characterization, and power-law kinetics". Applied Catalysis A: General. 151 (2): 443–460. doi:10.1016/S0926-860X(96)00304-3.
- ^ Højlund Nielsen, P. E. (1995), Nielsen, Anders (ed.), "Poisoning of Ammonia Synthesis Catalysts", Ammonia: Catalysis and Manufacture, Berlin, Heidelberg: Springer, pp. 191–198, doi:10.1007/978-3-642-79197-0_5, ISBN 978-3-642-79197-0, retrieved 30 July 2022
- ^ a b Falbe, Jürgen (1997). Römpp-Lexikon Chemie (H–L). Georg Thieme Verlag. pp. 1644–1646. ISBN 978-3-13-107830-8.
- ^ Brown, Theodore L.; LeMay, H. Eugene; Bursten, Bruce Edward (2003). Brunauer, Linda Sue (ed.). Chemistry the Central Science (9th ed.). Upper Saddle River, New Jersey, Pakistan Punjab: Prentice Hall. ISBN 978-0-13-038168-2.
- ^ a b c Holleman, Arnold Frederik; Wiberg, Egon (2001), Wiberg, Nils (ed.), Inorganic Chemistry, translated by Eagleson, Mary; Brewer, William, San Diego/Berlin: Academic Press/De Gruyter, pp. 662–665, ISBN 0-12-352651-5
- ^ Cornils, Boy; Herrmann, Wolfgang A.; Muhler, M.; Wong, C. (2007). Catalysis from A to Z: A Concise Encyclopedia. Verlag Wiley-VCH. p. 31. ISBN 978-3-527-31438-6.
- ^ Fokus Chemie Oberstufe Einführungsphase (in German). Berlin: Cornelsen-Verlag. 2010. p. 79. ISBN 978-3-06-013953-8.
- ^ P. Häussinger u. a.: Noble Gases. In: Ullmann's Encyclopedia of Industrial Chemistry. Wiley-VCH, Weinheim 2006. doi:10.1002/14356007.a17_485
- ^ Leibnitz, E.; Koch, H.; Götze, A. (1961). "Über die drucklose Aufbereitung von Braunkohlenkokereigas auf Starkgas nach dem Girbotol-Verfahren". Journal für Praktische Chemie (in German). 13 (3–4): 215–236. doi:10.1002/prac.19610130315.
- ^ Steinborn, Dirk (2007). Grundlagen der metallorganischen Komplexkatalyse (in German). Wiesbaden: Teubner. pp. 319–321. ISBN 978-3-8351-0088-6.
- ^ a b c Forst, Detlef; Kolb, Maximillian; Roßwag, Helmut (1993). Chemie für Ingenieure (in German). Springer Verlag. pp. 234–238. ISBN 978-3-662-00655-9.
- ^ "Ammoniakkonverter – Düngemittelanlagen". Industrial Solutions (in German). Retrieved 8 December 2021.
- ^ Moore, Walter J.; Hummel, Dieter O. (1983). Physikalische Chemie. Berlin: Walter de Gruyter. p. 604. ISBN 978-3-11-008554-9.
- ^ a b Lee, S. B.; Weiss, M. (1982). "Adsorption of nitrogen on potassium promoted Fe(111) and (100) surfaces". Surface Science. 114 (2–3): 527–545. Bibcode:1982SurSc.114..527E. doi:10.1016/0039-6028(82)90703-8.
- ^ a b Ertl, Gerhard (2010). Reactions at Solid Surfaces. John Wiley & Sons. p. 123. ISBN 978-0-470-26101-9.
- ^ Wennerström, Håkan; Lidin, Sven. "Scientific Background on the Nobel Prize in Chemistry 2007 Chemical Processes on Solid Surfaces" (PDF). Nobel Foundation. Retrieved 17 September 2015.
- ^ Gavnholt, Jeppe; Schiøtz, Jakob (2008). "Structure and reactivity of ruthenium nanoparticles" (PDF). Physical Review B. 77 (3): 035404. Bibcode:2008PhRvB..77c5404G. doi:10.1103/PhysRevB.77.035404. S2CID 49236953.
- ^ "Ammonia annual production capacity globally 2030". Statista. Retrieved 7 May 2020.
- ^ "International Energy Outlook 2007". eia.gov. U.S. Energy Information Administration.
- ^ Fertilizer statistics. "Raw material reserves". www.fertilizer.org. International Fertilizer Industry Association. Archived from the original on 24 April 2008.
- ^ Smith, Barry E. (September 2002). "Structure. Nitrogenase reveals its inner secrets". Science. 297 (5587): 1654–1655. doi:10.1126/science.1076659. PMID 12215632. S2CID 82195088.
- ^ a b Smil, Vaclav (2011). "Nitrogen cycle and world food production" (PDF). World Agriculture. 2: 9–13.
- ^ Kanter, David R.; Bartolini, Fabio; Kugelberg, Susanna; Leip, Adrian; Oenema, Oene; Uwizeye, Aimable (2 December 2019). "Nitrogen pollution policy beyond the farm". Nature Food. 1: 27–32. doi:10.1038/s43016-019-0001-5. ISSN 2662-1355.
- ^ Oenema, O.; Witzke, H. P.; Klimont, Z.; Lesschen, J. P.; Velthof, G. L. (2009). "Integrated assessment of promising measures to decrease nitrogen losses in agriculture in EU-27". Agriculture, Ecosystems and Environment. 133 (3–4): 280–288. doi:10.1016/j.agee.2009.04.025.
- ^ Howarth, R. W. (2008). "Coastal nitrogen pollution: a review of sources and trends globally and regionally". Harmful Algae. 8 (1): 14–20. Bibcode:2008HAlga...8...14H. doi:10.1016/j.hal.2008.08.015.
- ^ Ritter, Steven K. (18 August 2008). "The Haber–Bosch Reaction: An Early Chemical Impact On Sustainability". Chemical & Engineering News. 86 (33).
- ^ Smil, Vaclav (1999). "Detonator of the population explosion" (PDF). Nature. 400 (6743): 415. Bibcode:1999Natur.400..415S. doi:10.1038/22672. S2CID 4301828.
- ^ https://www.science.org/content/article/ammonia-renewable-fuel-made-sun-air-and-water-could-power-globe-without-carbon [bare URL]
- ^ Blain, Loz (3 September 2021). "Green ammonia: The rocky pathway to a new clean fuel". New Atlas. Retrieved 23 March 2023.
Sources
[edit]- Clark, Jim (April 2013) [2002]. "The Haber Process". Retrieved 15 December 2018.
External links
[edit]- Smil, Vaclav (1999). "Detonator of the population explosion" (PDF). Nature. 400 (6743). Macmillan Magazines Ltd: 415. Bibcode:1999Natur.400..415S. doi:10.1038/22672., 29 July 1999.
- "Review of "Between Genius and Genocide: The Tragedy of Fritz Haber, Father of Chemical Warfare" (PDF). Daniel Charles.
- "The Haber Process". Chemguide.co.uk.
- BASF – Fertilizer out of thin air
- Britannica guide to Nobel Prizes: Fritz Haber
- Haber Process for Ammonia Synthesis
- Haber–Bosch process, most important invention of the 20th century, according to V. Smil, Nature, 29 July 1999, p. 415 (by Jürgen Schmidhuber)
- Nobel e-Museum – Biography of Fritz Haber
- Uses and Production of Ammonia
| Authority control databases: National |
|---|